erektion — ejakulation — penis — hoden — endokrinologie — fertilität — genetische aspekte — aging male — varia
Genetische Ursachen einer defekten gonadotropen Achse
Eine Unterfunktion der Hoden wird allgemein als Hypogonadismus bezeichnet. Die hieraus resultierende Symptomatik ist einerseits vom
Ausmaß des Testosteronmangels und andererseits vom Zeitpunkt der Manifestation geprägt. Beim sekundären Hypogonadismus liegt eine Störung
der übergeordneten Regulationszentren im Hypothalamus und/oder Hypophyse vor. Diese ist durch eine fehlende oder erniedrigte
Gonadotropin-
Beim idiopathischen hypogonadotropen Hypogonadismus wird zwischen den im Zusammenhang mit An-/Hyposmie auftretenden und isolierten Formen unterschieden
-
Dem idiopathischen hypogonadotropen Hypogonadismus liegt eine mangelnde bzw. fehlende hypothalamische Gonadotropin-
-
Die Verbindung von fehlendem oder vermindertem Geruchssinn und Hypogonadismus wurde erstmals von dem Psychologen Franz Josef Kallmann
beschrieben, der zugleich Namensgeber des damit verbundenen Symptomenkomplexes wurde. Ursächlich ist eine Agenesie des Bulbus olfactorius,
durch die GnRH-
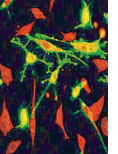
|
Abb. 1: Onthogenetisch werden die GnRH-Neuronen ausserhalb des Gehirns im nasalen Kompartiment in der Umgebung der olfaktorischen Plakode angelegt. Bei ihrer Wanderung via Siebbeinplatte des Os ethmoidale in den mediobasalen Hypothalamus bedienen sie sich einer Art Führungsschiene, die ihnen in Forn von Riechnervenfasern zur Verfügung steht. Dem Kallmann-Syndrom liegt eine Agenesie des Bulbus olfactorius zugrunde, so dass die wandernden GnRH-Neuronen im Bereich der Siebbeinplatte liegen bleiben. |
Bei Patienten mit Kallmann-Syndrom wurde erstmals eine molekulare Grundlage für ererbten idiopathischen hypogonadotropen Hypogonadismus aufgeklärt. Diese Fälle ließen sich auf Mutationen im X-chromosomal rezessiv vererbten KAL1-Gen zurückführen. Das KAL1-
Genetische Untersuchungen an insgesamt 109 Patienten mit idiopatischem hypogonadotropen Hypogonadismus ergaben nur in vier Fällen (3,7%)
eine Mutation im KAL1-Gen. Alle diese Fälle betrafen 63 der 109 Männer mit An-/Hyposmie, so dass sich die Prävalenz für diese Gruppe auf 6,3%
erhöhte. Damit ergibt sich die Folgerung, dass KAL1-
Seit Kurzem zählen auch die Gene von Prokineticin-2 (PROK2) und dem Prokineticin-Rezeptor-2 (PROKR2) zu den Kandidaten, bei denen
Mutationen an der Pathogenese des Kallmann-
Untersuchungen an Mäusen, aber auch Daten eines Mutationsscreenings bei Kallmann-Patienten, haben Indizien für eine Rolle des nasalen
mbryonalen luteinisierendes-
-
Isolierte, nicht mit An-/Hyposmie im Zusammenhang stehende Formen eines hypogonadotropen Hypogonadismus treten sowohl sporadisch als auch genetisch
bedingt auf. Letztere sind in knapp 50% der Fälle auf Mutationen im GnRH-Rezeptor zurückzuführen. Es kommt eine Reihe von compound heterozygoten
(unterschiedliche Mutationen in beiden mutierten Allelen eines Gens) und homozygoten Mutationen des GnRH-Rezeptors vor [8]. Bewirkt wird eine Störung
der Gonadotropinsekretion auf hypophysärer Ebene, mit der Folge eines partiellen oder vollständigen Hypogonadismus. Frequenz und Amplitude der
pulsatilen LH-Freisetzung sind reduziert.
Mutationen im G-Protein-gekoppelten Rezeptor 54 (GPR54) sind eine weitere, erst 2003 entdeckte Ursache für einen isolierten hypogonadotropen
Hypogonadismus [9]. Als natürlicher Ligand für GPR54 fungiert Kisspeptin, das im basalen Vorderhirn gebildet wird und bei der
Steroidhormon-
Aktuell wurde eine Mutation des FGFR1-Gens in einer Familie beschrieben, bei der kein hypogonadotroper Hypogonadismus in Form eines Kallmann-Syndroms –
wie bei Mutationen dieses Gens üblich – ausgeprägt ist, sondern ein normosmischer, isolierter hypogonadotroper Hypogonadismus besteht. Der Unterschied
zum Kallmann-
Abb. 1: Unterhalb bestimmter Schwellenwerte der Testosteronkonzentration traten verschiedene Symptome im Patientenkollektiv jeweils
signifikant gehäuft auf (n = Anzahl der Patienten je Sechstile der gemessenen Testosteronkonzentrationen) [nach 5].
-
Bei keinem der Genotypen an einem für idiopathischen hypogonadotropen Hypogonadismus oder das Kallmann-Syndrom identifizierten Genorte
lässt sich verlässlich auf den manifestierten Phänotyp schließen. Sowohl innerhalb betroffener Familien als auch interfamiliär ist die
jeweilige Krankheit zum Teil sehr variabel ausgeprägt. Das lässt Zweifel an deren Monogenizität aufkommen. Vielmehr ist davon auszugehen,
dass bei idiopathischem hypogonadotropen Hypogonadismus oder Kallmann-Syndrom oft mehr als ein mutiertes Gen involviert ist.
Starke Indizien für diese Hypothese lieferten jüngst publizierte Ergebnisse der Untersuchungen an zwei Familien: In einer Familie kommt das
Kallmann-Syndrom vor und in der anderen ein normosmischer hypogonadotroper Hypogonadismus in jeweils erheblich variabler Ausprägung vor.
Für das Kallmann-Syndrom war eine heterozygote FGFR1-Mutation und für den normosmischen hypogonadotropen Hypogonadismus eine compound
heterozygote Mutation im Gen für den GnRH-Rezeptor nachgewiesen worden. Bei ergänzenden Untersuchungen wurden in Familie 1 noch eine
Deletion im NELF-Gen und in Familie 2 eine heterozygote FGFR1-Mutation identifiziert. Daraus wird geschlossen, dass zwei unterschiedliche
Gendefekte synergistisch zusammenwirken und einen schwereren Phänotyp in Familien mit idiopathischem hypogonadotropen Hypogonadismus oder
Kallmann-Syndrom bewirken können als jeder der beiden Gendefekte für sich [12].
|
Wichtig ist das frühzeitige Erkennen eines hypogonadotropen Hypogonadismus und die rechtzeitige Einleitung einer Testosteron- |
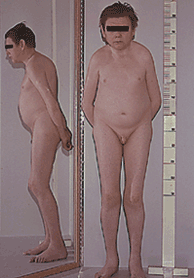 33-jähriger Patient mit idiopathischem hypogonadotropen Hypogonadismus, der bis zu diesem Zeitpunkt nicht substituiert worden war. Es hatte sich eine manifeste Osteoporose mit Wirbelkörperdeformierungen (Fisch- und Keilwirbelbildungen und Kompressionsfrakturen) entwickelt, so dass der Patient invalidisiert wurde. Charakteristisch ist der eunuchoide Hochwuchs mit relativ langen Extremitäten und kurzem Rumpf sowie der ausgeprägte Rundrücken. Auffallend sind auch die nur schwach ausgebildete Muskulatur bei schlaffer Körperhaltung und die pigmentarme, vorgealterte Haut. (Foto: F. Husmann) | |
|
Patienten mit einem hypogonado-tropen Hypogonadismus leiden massiv unter einem Testosteron-
Ein eunuchoider Hochwuchs lässt sich naturgemäß nicht korrigieren. Dass es dazu kommt kann aber verhindert werden, sofern die Diagnose rechtzeitig erfolgt
und unverzüglich mit einer Substitution von Testoste- |
der Muskulatur. Da Testosteron auch bei der Aktivierung des Erythropoe-
Besteht bei Patienten mit idiopathi- | |
Bei Maldeszensus, Mikropenis und gestörter GnRH-Sekretion frühzeitiger Verdacht auf Prader-Labhart-Willi-Syndrom
-
Das erstmals 1956 beschriebene Prader-Labhart-Willi-Syndrom (Prävalenz: 1:15.00025.000) ist durch einen hypogonadotropen Hypogonadismus
kombiniert mit Hypotonie der Muskulatur, Minderwuchs, Oligophrenie, faziale Dysmorphie, Adipositas und Diabetes mellitus Typ 2 gekennzeichnet.
Darüber hinaus finden sich oft eine Akromikrie, Strabismus, Skoliose und Zahnschmelzdefekte. Als Ursache gilt eine väterlicherseits vererbte
Deletion des langen Arms des Chromosoms 15. Eine Verdachtsdiagnose kann aufgrund eines Maldeszensus und eines Mikropenis sowie gestörter
GnRH-Sekretion oft schon bei der Geburt gestellt werden.
Neuere Befunde weisen darauf hin, dass ein hypothalamischer Defekt wohl nicht die einzige Ursache des Prader-
-
Das Bardet-Biedl-Syndrom ist eine früher mit dem Laurence-Moon-Syndrom zusammengefasste autosomal rezessive Krankheit, die durch
abdominale Adipositas, mentale Retardierung, dysmorphe Extremitäten, Retinadystrophie bzw. pigmentäre Retinopathie, Nierenanomalien
und in einigen Fällen Hypogonadismus bzw. Hypogenitalismus (letztere nur beim männlichen Geschlecht) gekennzeichnet ist [13].
Ursprünglich bestand die Auffassung, dass diese Syndrome ebenfalls mit einem hypogonadotropen Hypogonadismus in Verbindung stehen.
Neueren Befunden ist der Hypogonadismus in diesem Zusammenhang aber nicht hypothalamisch-hypophysärer Natur. Dennoch gilt es diese sehr
seltene Krankheit gegenüber ähnlichen Syndromen differnzialdiagnostisch abzuklären (Abb. 2).
-
Beim Pasqualini-Syndrom finden sich bei normalem FSH-Spiegel erniedrigte LH- und Testosteronspiegel im Serum. Es handelt es sich
um eine anlagebedingte isolierte Sekretionsstörung des LH, d.h. eine Form des kongenitalen isolierten hypogonadotropen Hypogonadismus.
In der Klinik fallen diesbezügliche Patienten durch eine unzureichende Virilisierung bei normal großen Hoden auf. Trivial wird auch vom
Syndrom der fertilen Eunuchen gesprochen.
Literatur:
[1] Simoni M, Nieschlag E. 2007. Genetics of hypogonadotropic hypogonadism. Horm Res 67(suppl 1):149-154.
[2] Karges B, de Roux N. 2005. Molecular genetics of isolated hypogonadotropic hypogonadism and Kallmann syndrome. Endocr Dev 8;67-80.
[3] Bhagavath B, Xu N, Ozata M, et al. 2007. KAL1 mutations are not a common cause of idiopathic hypogonadotrophic hypogonadism in humans. Mol Hum Reprod 13:25-30.
[4] Dodé C, Levilliers J, Dupont JM, et al. 2003. Loss-of-function mutations in FGFR1 cause autosomal dominant Kallmann syndrome. Nat Genet 33:463-465.
[5] Dodé C, Teixeira L, Levilliers J, et al. 2006. Kallmann syndrome mutations in the genes encoding prokineticin-2 and prokineticin receptor-2. PLoS Genet 2:e175.
[6] Matsumoto S, Yamazaki C, Matsumoto KH, et al. 2006. Abnormal development of the olfactory bulb and reproductive system in mice lacking prokineticin receptor PKR2. Proc Natl Acad Sci USA 103:4140-4145.
[7] Miura K, Acierno JS Jr, Seminara SB. 2004. Characterization of the human nasal embryonic LHRH factor gene, NELF, and a mutation screening among 65 patients with idiopathic hypogonadotropic hypogonadism (Hypergonadotroper Hypogonadismus). J Hum Genet 49:265-268.
[8] Lanfranco F, Gromoll J, von Eckardstein S, et al. 2005. Role of sequence variations of the GnRH receptor and G protein-coupled receptor 54 gene in male idiopathic hypogonadotropic hypogonadism. Eur J Endocrinol 153:845-852.
[9] de Roux N, Genin E, Carel JC, et al. 2003. Hypogonadotropic Hypogonadism due to loss of function of KISS1-derived peptide receptor GPR54. Proc Natl Acad Sci USA 100:10972-10976.
[10] Xu N, Qin Y, Reindollar RH, et al. 2007. A mutation in the fibroblast growth factor receptor 1 gene causes fully penetrant normosmic isolated hypogonadotropic hypogonadism. J Clin Endocrinol Metab 92:1155-1158.
[11] Pitteloud N, Quinton R, Pearce S, et al. 2007. Digenic mutations account for variable phenotypes in idiopathic hypogonadotropic hypogonadism. J Clin Invest 17:457-463.
[12] Eiholzer U, l´Allemand D, Rousson V, et al. 2006. Hypothalamic and gonadal components of hypogonadism in boys with Prader-Labhart-Willi syndrome. J Clin Endocrinol Metab 91:892-898.
[13] Ianello S, Bosco P, Cavaleri A, et al. 2002. A review of the literature of Bardet-Biedl disease of the cases associated with metabolic syndrome and diagnosed after age of fifty. Obes Res 3:123-135.
[14] Pitteloud N, Haves FJ, Dwyer A, et al. 2002. Predictors of outcome of long-term GnRH therapy in men with idiopathic hypogonadotropic hypogonadism. J Clin Endocrinol Metab 87:4128-4136.
© 2003-2026 pro-anima medizin medien
–
impressum
–
mediadaten
–
konzeption
–
datenschutz