erektion — ejakulation — penis — hoden — endokrinologie — fertilität — genetische aspekte — aging male — varia
Mutiertes Phosphatase-Gen als eine Ursache identifiziert
Beim Noonan-Syndrom handelt es sich um eine genetische Erkrankung, die hauptsächlich durch unterschiedlich
stark ausgeprägte dysmorphe Gesichtszüge, einen proportionierten Kleinwuchs, gelegentliche Pterygiumbildung,
Fehlbildungen innerer Organe wie insbesondere des Herzens und einen Maldescensus testis mit häufig auftretenden
Fertilitätsstörungen gekennzeichnet ist. Diese Entwicklungsstörung tritt mit einer Prävalenz von 1:2.500
bis 1:1.000 relativ häufig auf. Ihre Pathogenese ist noch weitgehend unklar. Doch erst kürzlich ist es gelungen,
ein Gen dingfest zu machen, dessen Mutabilität unter anderem als Ursache der kardialen Fehlbildungen beim
Noonan-
Es gibt kein männliches Turner-Syndrom
-
Verschiedene klinische Merkmale beim Noonan-Syndrom erinnern an das Ullrich-Turner-Syndrom, so dass ursprünglich
die irreführenden Bezeichnungen männliches Turner-Syndrom oder weibliches Pseudo-Turner-Syndrom gebräuchlich
waren. Doch anders als beim Ullrich-Turner-Sydrom handelt es sich beim Noonan-Syndrom weder um eine zahlenmäßige,
noch um eine strukturelle Chromosomenanomalie. Vielmehr liegt eine autosomal-dominant vererbliche, genetische
Entwicklungsstörung vor.
Das Noonan-Syndrom tritt sporadisch oder familiär auf und betrifft beide Geschlechter gleichermaßen. Die Diagnose ließ sich bis vor kurzem ausschließlich klinisch stellen.
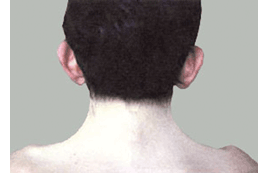
Das äußere Erscheinungsbild beim Noonan-Syndrom weist charakteristische Merkmale im Gesichtsbereich auf. Typisch sind eine breite Stirn, weit auseinander liegende, schräg stehende Augen, hängende Oberlider und breite Nasenflügel. Charakteristische Hinweise liefern ferner ein kurzer Hals und eine Pterygiumbildung (Abb.). Bei Neugeborenen ist der Nackenbereich durch überschüssige Haut oft in Falten gelegt.
Berichtet wird von einer geringen bis mittelgradigen Retardierung. Eine solche kann möglicherweise aber auch
durch das Wahrnehmen der Andersartigkeit begünstigt sein.
-
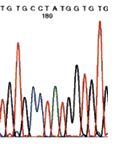
Bereits vor knapp zehn Jahren ist es gelungen, den Sitz für ein vermutetes ursächliches Gen auf dem langen Arm
des Chromosoms 12 einzukreisen. Zugleich wurde aber auch erkannt, dass beim Noonan-
Verschiedene Arbeitsgruppen haben kürzlich ein Gen (PTPN11) auf dem Chromosom 12 identifiziert, das bei 30% bis 50% der
daraufhin untersuchten Noonan-
Intrazelluläre Phosphatasen sind zumeist spezialisierte Enzyme, die Phosphatreste von bestimmten funktionellen Proteinen
abspalten. Sie beeinflussen dadurch die Dauer des aktiven Zustands solcher Proteine. Darüber hinaus wechseln diese Phosphatasen
selbst zwischen einem aktiven und einem inaktiven Zustand hin und her. Bei den identifizierten Mutationen von SHP-2 tritt
jeweils ein funktioneller Zugewinn ein (gain of function), d.h. sie sind fortwährend aktiv.
Die Entdeckung von Mutatio-nen im PTPN11-Gen bei einem Teil der Patienten mit Noonan-Syndrom, rückt diese Entwicklungsstörung
in den Kreis von Krankheiten, deren phänotypische und klinische Diagnose durch eine genetische Analyse abgesichert werden kann.
Hiervon verspricht man sich eine Optimierung der therapeutischen Vorgehensweise. Unter anderem gibt es Beobachtungen, dass
im Wachstum zurückbleibende Jugendliche mit einem Noonan-
-
Sowohl bei Jungen als auch Mädchen mit Noonan-Syndrom tritt die Pubertät zumeist verzögert ein. Therapeutisch kann hierbei
in gleicher Weise wie bei der konstitutionellen Pubertas retarda vorgegangen werden.
Während bei Mädchen die weitere Geschlechtsentwicklung meist regelhaft verläuft, kommt es bei Jungen in etwa 60% der Fälle
zu einer Hodendeszensusstörung. Als Folge hiervon bestehen bei einem Großteil der Noonan-Patienten Fertilitätsstörungen.
Man findet teilweise Männer mit Oligozoospermie oder Azoospermie. Ferner ist die Testosteronproduktion durch die Lageanomalie
der Hoden unter Umständen eingeschränkt. Es empfiehlt sich daher die Lageanomalie der Hoden bereits frühzeitig zu korrigieren.
Literatur:
[1] Tartaglia M, Mehler EL, Goldberg R, et al. 2001.
Mutations in PTPN11, encoding the protein tyrosine phosphatase SHP-2, cause Noonan syndrome. Nat Genet 29:465-468.
[2] Tartaglia M, Kalidas K, Shaw A, et al. 2002. PTPN11 mutations in Noonan syndrome: molecular spectrum,
genotype-phenotype correlation, and phenotypic heterogeneity. Am J Hum Genet 70:1555-1563.
[3] Kosaki K, Suzuki T, Muroya K, et al. 2002.
PTPN11 (Protein-tyrosine phosphatase, nonreceptor-type 11) mutations in seven Japanese patients with Noonan syndrome.
J Clin Endocrinol Metab 87:3529-3533.
© 2003-2026 pro-anima medizin medien
–
impressum
–
mediadaten
–
konzeption
–
datenschutz