erektion — ejakulation — penis — hoden — endokrinologie — fertilität — genetische aspekte — aging male — varia
Genetische Ursachen der olfakto-genitalen Störung
Bereits in Jahr 1856 waren Maestre de San Juan bei der Autopsie eines 40-jährigen Mannes dessen sehr kleine Hoden, Mikropenis und fehlende Riechkolben aufgefallen. Fast ein Jahrhundert später berichtete im Jahre 1944 der in Deutschland geborene amerikanische Psychiater Franz Josef Kallmann über drei Familien, bei deren Mitgliedern gehäuft eunuchoider Habitus und fehlender Geruchssinn gemeinsam vorkamen. Die offensichtliche Vererbbarkeit dieser Merkmale ließ auf eine genetische Ätiologie schließen.
Als man später in der Lage war, im Urin Gonadotropine zu bestimmen, wurde die gonadale Komponente des Kallmann
Syndroms als eine Form des hypogonadotropen Hypogonadismus erkannt. Nach heutigem Kenntnisstand beruht dieser auf
einem angeborenen Defekt der Gonadotropin-
Beim Kallmann-Syndrom handelt es sich um eine selten vorkommende genetische Störung, deren Prävalenz oft nicht
getrennt von Fällen des isolierten idiopathischen hypogonadotropen Hypogonadismus (IHH) ermittelt wurde. Neueren
Angaben zufolge tritt das Kallmann-Syndrom bei einem von 8.000 Männern auf, während das Verhältnis bei den
Frauen 1:40.000 beträgt [1]. Dieses ungleich auf beide Geschlechter verteilte Erkrankungsrisiko
gab lange Zeit Rätsel auf und findet wie am Ende des Artikels dargelegt wird erst durch neueste
Forschungsergebnisse eine hypothetische Erklärung.
Die diagnostische Abgrenzung des Kallmann-Syndroms bzw. des IHH gegenüber einer konstitutionellen Entwicklungsstörung
gestaltet sich schwierig. Nachdem zunächst deutlich subnormale Werte an FSH, LH und Testosteron ermittelt wurden, kann
zur differentiellen Abklärung ein GnRH-Test (100 mg) nach vorausgegangener 36-stündiger Stimulation mit pulsatilem
GnRH gemacht werden. Beim Kallmann-
Kernpunkt der Behandlung beim Kallmann-Syndrom ist der lebenslange Ausgleich des Testosteronmangels, durch den
insbesondere das Osteoporoserisiko gesenkt wird.
Männer mit Kallmann-Syndrom sind infertil. Doch läßt sich Fertilität mit gutem Erfolg durch eine Behandlung mit
Gonadotropinen oder mit pulsatilen GnRH-Gaben herstellen. Bei einigen Patienten kann es allerdings zwei Jahre oder
länger dauern, bis die volle Spermienproduktion in Gang kommt.
Im Hypothalamus verteilen sich die nur etwa 1.000 GnRH-Neuronen, ohne sich in einer Kerngruppe zu organisieren.
Trotzdem bilden sie in ihrer Gesamtheit den GnRH-
Aber auch innerhalb einer miteinander verschwägerten Sippe können einzelne Personen das klassische Kallmann-
Die Mehrheit der daraufhin untersuchten Fälle von familiärem Kallmann-
Anosmine haben sich im Lauf der Evolution kaum verändert. Daher kann man sogar an Invertebraten wie der Nematode
Caenorhabditis elegans (Abb.) am homologen Gen von KAL-1 untersuchen, wie sich Mutationen auf neuronales
Wachstum auswirken [4].
Obwohl die Entstehung der olfakto-genitalen Symptomatik beim Kallmann-<
Die Verbindung zum Kallmann-Syndrom ergibt sich aus Hinweisen, dass Anosmin-1, das Gen-Produkt von KAL-1, für die Reaktion von
FGF mit dem FGF-Rezeptor-1 erforderlich ist. Darüber hinaus wird angenommen, dass sich KAL-1 partiell der X-Inaktivierung entzieht
und somit bei Frauen mehr Anosmin-1 als bei Männern bebildet wird. Das wäre auch eine Erklärung für die erhöhte Prävalenz des
Kallmann-
Männer häufiger betroffen als Frauen
Das Kallmann-Syndrom ist per Definition ein hypogonadotroper Hypogonadismus, der in Verbindung mit Anosmie bzw.
Hyposmie auftritt. Allerdings kann die Ausprägung der beiden Merkmale sehr variabel sein und vielfach kommen
weitere neurologische und nicht neurologische Störungen hinzu.
Ausbleibende pubertäre Entwicklung als Leitsymptom
Meist werden Knaben mit Kallmann-Syndrom erst auffällig, wenn die Pubertät auf sich warten lässt oder die
sexuelle Entwicklung unvollständig bleibt. Als Ursache hierfür ist das Ausbleiben der GnRH-Sekretion aus
dem Hypothalamus erkannt worden. Wird die Krankheit erst nach dem 16. Lebensjahr erkannt, haben sich aufgrund
des Hypogonadismus zumeist bereits eunuchoide Skelettproportionen herausgebildet.
Migationsdefekt während der Embryonalentwicklung
Während der frühen Embryonalentwicklung werden die GnRH-Neuronen im Epithel der Riechplakode angelegt und wandern
von dort aus entlang der zentralen Fortsätze des Nervus terminalis ins Gehirn. Auf der Migrationsstrecke lassen
sich die Nervenfasern mit Antikörpern gegen Zelladhäsionsmoleküle markieren [2].
Beträchtliche genotypische und phänotypische Heterogenität
Das Kallmann-Syndrom tritt mehrheitlich sporadisch und mit einer erheblich geringeren Inzidenz auch familiär auf.
Die involvierten Gene sind X-gekoppelt, autosomal dominant und autosomal rezessiv, so dass von einer erheblichen
genotypischen und phänotypischen Heterogenität auszugehen ist.
X-Chromosomale Form des Kallmann-Syndroms
Für die X-gekoppelte Form des Kallmann-Syndroms wird das Gen KAL-1 verantwortlich gemacht. Das zugehörige Proteinprodukt
Anosmin-1 ist ein extrazelluläres Matrixprotein, das nach Sequenzanalysen vermutlich eine Domäne besitzt, die als
Proteinase-
Autosomal dominante Form des Kallmann-Syndroms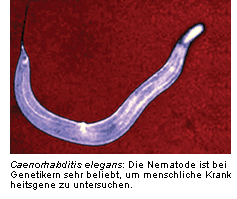
Neuerdings gibt es Hinweise darauf, dass der Fibroblasten-Wachstumsfaktor (FGF) bei der Ausbildung eines Kallmann-
Literatur:
[1] Hu Y, Tanriverdi F, MacColl GS, Bouloux P-MG. 2003.
Kallmann´s syndrome: molecular pathogenesis. Int J Biochem Cell Biol 35:1157-1162.
[2] Schwanzel-Fukuda M, Crossin KL, Pfaff DW, et al. 1996.
Migration of luteinizing hormone-releasing hormone (LHRH) neurons in early human embryos. J Comp Neurol 366:547-557.
[3] Oliveira LMB, Seminara SB, Beranova M, et al. 2001.
The importance of autosomal genes in Kallmann syndrome: genotype-phenotype correlations and neuro-endocrine characteristics.
J Clin Endocrinol Metab 86:1532-1538.
[4] Rugarli EI, Schiavi ED, Hilliard MA, et al. 2003.
The Kallmann syndrome gene homolog in C. elegans is involved in epidermal morphogenesis and neurite branching. Development 129:1283-1294.
[5] Dode C, Levilliers J, Dupont JM, et al. 2003.
Loss-of-function mutation in FGFR1 cause autosomal dominant Kallmann syndrome. Nat Genet 33:463-465.
© 2003-2024 pro-anima medizin medien
–
impressum
–
mediadaten
–
konzeption
–
datenschutz